In our last post, we discussed some of the news from the American Society of Hematology (ASH) meeting in December on advances in the treatment of hematological cancers. In this second post, we look at new therapeutic developments highlighted at ASH for hemophilia, beta thalassemia and sickle cell disease.
Therapies for Hemophilia
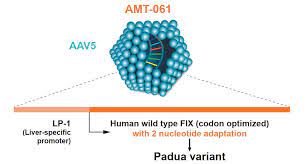
UniQure and their partner, CSL Behring, presented positive results from the HOPE-B pivotal Phase 3 trial of their gene therapy, AMT-061 (Etranacogene dezaparvovec) in patients with severe to moderately severe hemophilia B, which is caused by a deficiency in clotting factor IX (FIX). AMT-061 is designed to deliver a highly functional copy of the F9 gene (FIX-Padua) to patients’ cells, which produces FIX clotting activity eight times greater than that of the standard F9 gene. The companies announced that the trial met the primary endpoint of non-inferiority in annualized bleeding rate (ABR), assessed 18-months following a single administration of the therapy, compared to baseline FIX prophylactic therapy. The gene therapy also achieved the trial’s secondary endpoint by demonstrating statistical superiority in reducing treated patients’ ABR compared to baseline FIX prophylactic therapy. The benefit produced by the gene therapy was stable and durable, with a mean FIX activity of 39.0 percent of normal at 6 months and 36.9 percent of normal at 18 months in the full study population. A single intravenous dose of AMT-061 appears to offer a functional cure for patients with severe hemophilia B, improving their condition from a severe to a mild impairment or normal function of FIX activity. UniQure and CSL Behring plan to seek marketing approval for their hemophilia B gene therapy in both the United States and European Union in the first half of 2022.
BioMarin is developing a gene therapy, Roctavian (valoctocogene roxaparvovec), for hemophilia A, caused by a deficiency of clotting Factor VIII. The FDA rejected BioMarin’s treatment in 2020, requesting additional clinical data which the company has since been amassing through a global Phase III trial. In January 2022, BioMarin announced two-year results from the study, showing that treated patients experienced an 85% reduction from baseline in annualized bleeding rate (demonstrating superiority to Factor VIII prophylaxis) and a 98% reduction from baseline in annualized Factor VIII infusions. At the same time, Factor VIII activity for patients treated for at least three years appeared to lessen over time, raising the question of whether gene therapies could ever be one-and-done treatments in this indication. BioMarin’s Marketing Authorization Application is currently under review by the European Medicines Authority and the company expects to resubmit its application to the FDA later this year.
There are several other gene therapies for hemophilia at advanced development stages. For example, Pfizer is developing a hemophilia B gene therapy called fidanacogene elaparvovec, originally in partnership with Spark Therapeutics prior to their acquisition by Roche. The company expects to release initial results of a Phase 3 trial of this treatment in the first quarter of 2023.
Treatments for hemoglobinopathies: Beta-thalassemia
There was also promising gene therapy news at ASH for patients with transfusion-dependent beta thalassemia. Beta-thalassemia is an inherited blood disorder which in its most severe form results in the inability or near-inability to produce adult hemoglobin. This situation can lead to severe anemia and life-long dependence on red blood cell transfusions.
Bluebird bio presented long-term follow-up results (median of 32 months and up to 7 years post-treatment) from multiple studies showing that its betibeglogene autotemcel (beti-cel) gene therapy produced normal or near-normal hemoglobin levels and safely eliminated the need for transfusions in more than 90 percent of adult and pediatric patients. Beti-cel is a one-time gene therapy that adds functional copies of a modified form of the β-globin gene into a patient’s own hematopoietic stem cells (HSCs), giving the HSCs the potential to produce adult hemoglobin. A Biologic License Application for beti-cel is currently under FDA review, and a decision is expected in Q3 2022.
Treatments for hemoglobinopathies: Sickle Cell Disease
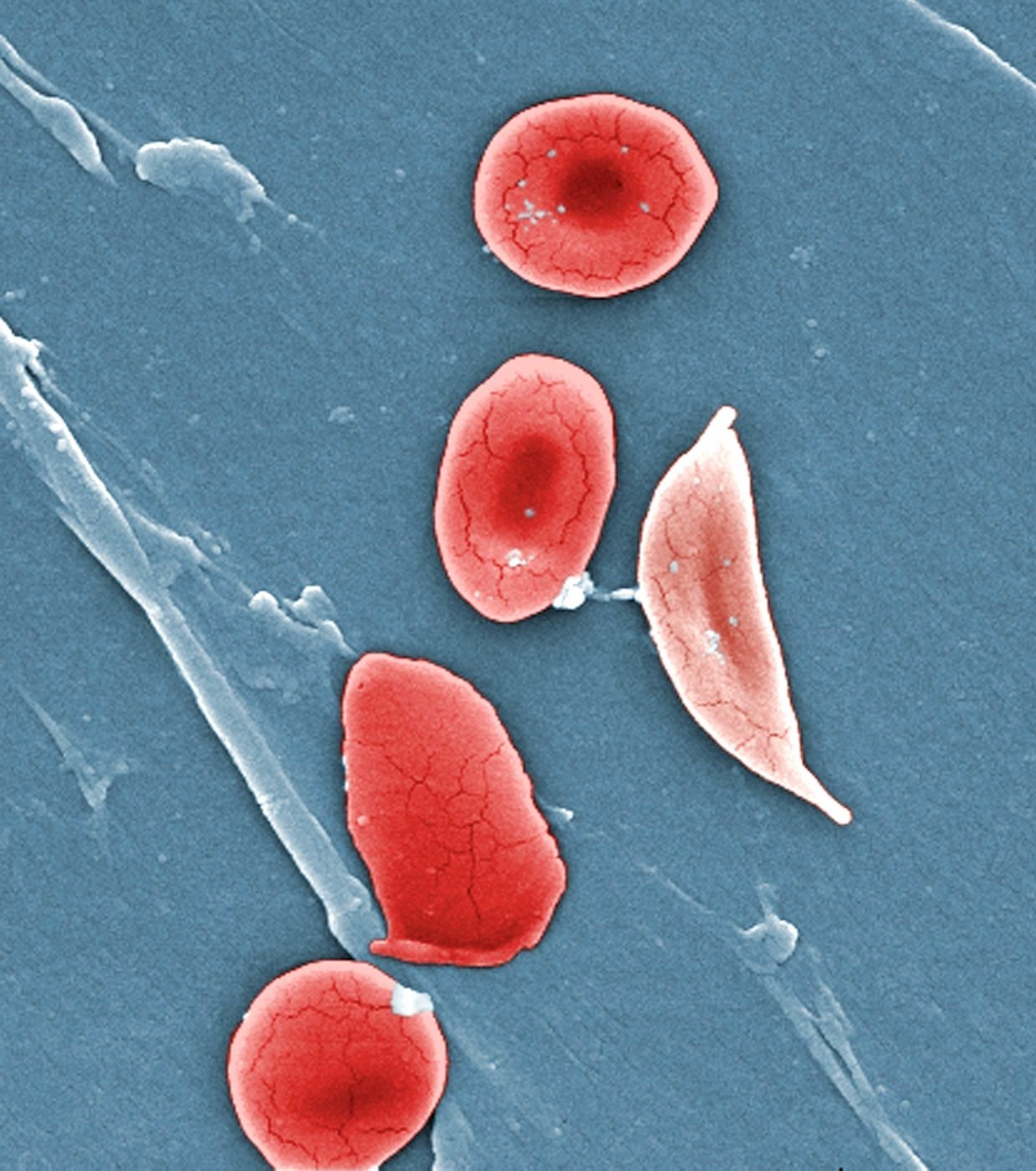
Bluebird also presented updated results from its Phase 1/2 study of lovotibeglogene autotemcel (lovo-cel) for sickle cell disease (SCD). SCD is an inherited, life-long hematological condition that affects millions of people worldwide, especially those with sub-Saharan African, Hispanic, South Asian, South European and Middle Eastern ancestry. The disease is characterized by hemolytic anemia, as well as painful vaso-occlusive crises resulting from the blockage of capillaries and small blood vessels by the sickled red blood cells (RBCs). This can lead to serious and often life-threatening complications including stroke, neurocognitive impairment, and organ damage.
Lovo-cel is a gene therapy designed to add functional copies of a modified form of the β-globin gene into a patient’s own HSCs, allowing their RBCs to produce anti-sickling hemoglobin, with the goal of reducing sickled RBCs, hemolysis, and other complications. In addition to reducing the incidence of painful vaso-occlusive events, which otherwise can occur multiple times per month, patients in a pivotal trial cohort achieved near-normal levels of key hemolysis markers and experienced sustained improvements in patient-reported quality of life following treatment.
While Vertex presented no new data at ASH, the company is also developing a potential one-and-done gene therapy with support from CRISPR Therapeutics. Their experimental treatment, CTX001, uses CRISPR-Cas9 to edit hematopoietic stem cells and produce healthy fetal hemoglobin rather than the deficient and faulty adult forms found in beta-thalassemia or sickle cell disease. Vertex is conducting a variety of Phase 1, 2, and 3 trials of CTX001 in both indications.
In the same area of therapeutics for SCD, Global Blood Therapeutics (GBT) presented data from multiple studies confirming the utility of their once-daily, oral treatment for hemolytic anemia, Oxbryta (voxelotor). The drug, first approved by the FDA in November 2019, directly inhibits sickle hemoglobin polymerization by increasing hemoglobin’s affinity for oxygen. At ASH, GBT presented results from multiple studies of the drug, including a retrospective analysis of real-world data from the Symphony Health Claims database on 2,700 SCD patients who were treated with Oxybryta between November 2019 and March 2021. Data from that analysis confirmed Oxbryta’s ability to produce a statistically significant improvement in blood hemoglobin levels and significant reductions in blood transfusions, vaso-occlusive crises, and hospitalizations.